It is helpful to have help files so make sure the path to your favorite web browser is set up. Press Continue.
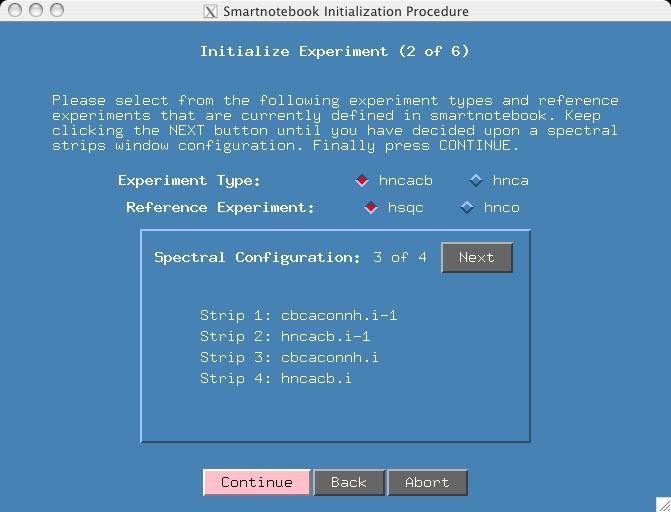
There are 4 spectral views and you can press the "Next" button to see them. Given that we have already seen the first configuration in the first example, please select "Spectral Configuration: 3 of 4". We will still use hsqc as the reference spectra. Press Continue.
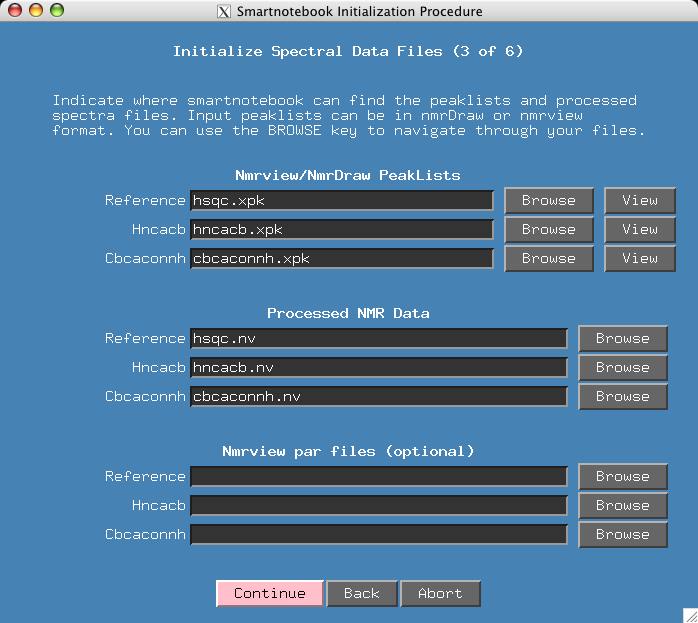
At this point we have a reasonably convenient window for entering peaklists and processed spectra. The default configuration for smartnotebook is H,C,N for x,y,z dimensions respectively. The processing also assumes that Ca will have a positive intensity and Cb will be negative.
It just so happens that I have chosen file names for the input data (in lower case) that match the title fields. Otherwise the user has to type the names of all the input files or use the "Browse" button. Press Continue.
If the user has Nmrview spectra parameter files (corresponding .par files for the .nv files) then these should be entered in the fields provided.
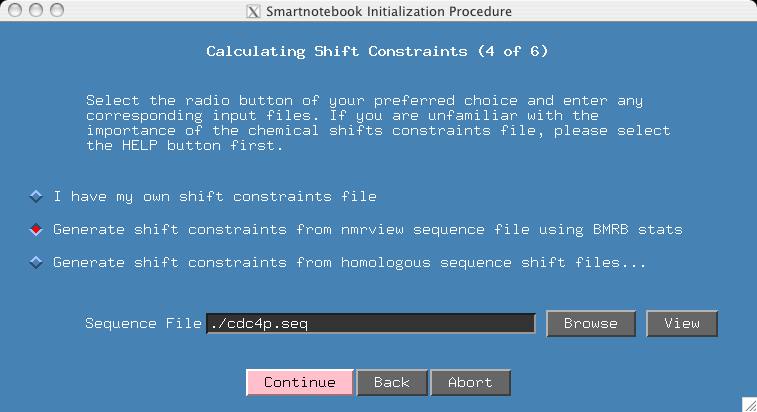
This section is more complex, so you may want to click the HELP button to know all your options. But for the purposes of this demo, we do not have the homologous shift files which would more tightly constrain expected shift values. If you have homologous shift data, then you will want to try the example presented later on in this section. If you select the middle radiobutton and if the name of your sequence input file ends in ".seq", then snb will be able to fill in the "Sequence File" entry. Press CONTINUE.
Here you enter the shifts constraints file. If smartnotebook has calculated it, then the field is automatically filled in with the filename. Press the View button to see what the file looks like.
This section deals with options. If you have customized colors and fonts for your smartnotebook environment, you may wish to import your own "AppDefaults" file. If you have an input ppm.out file where some atoms have been assigned, and you want the output ppm.out file to contain these assignments, then input your ppm.out file here. Please note that in the case of conflict between assignments made in snb and the input ppm.out file, the snb assignments will take precedence. Finally, it is possible you do not want snb to calculate connectivities because you plan to import your own (select the 'no' button in this case).
For most users, all you want to do is press "Finish Init". Expect this window to undergo many changes in upcoming versions.
Reading the following connectivity files. Found 0 connections in './snb.out/con.peakcon.dgg' Found 2144 connections in './snb.out/con.peakcon.dgx' Found 277 connections in './snb.out/con.peakcon.dxg' Found 1388 connections in './snb.out/con.peakcon.dxx'This is the end of this demo, if you exit smartnotebook now and then restart it you will see that we go directly to the smartnotebook main panel.