Search Button
This button activates a popup window which allows the
user to search for connectivities based on:
- Residue ID constraints
- Atom constraints
- Probability score
Example 1: I want to build a chain starting with a certain amino acid
type (eg serine).
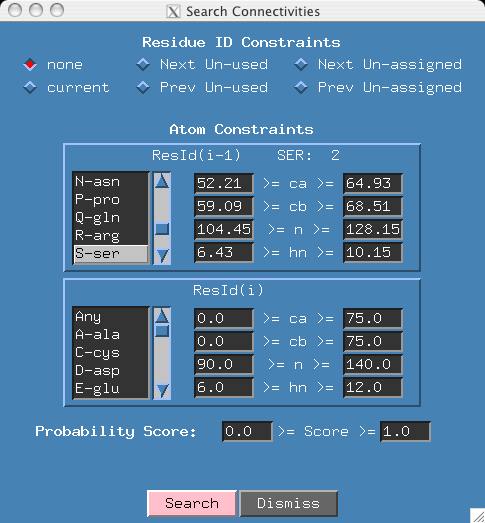
- Scroll the amino acid list of the ResID(i-1) atom constraints window and
click on the S-ser button. The software finds
the first SER in your sequence and places the associated atom constraints in
the boxes (eg 52.21 >= ca >= 64.93).
- For the amino acid list of ResID(i) click on "Any" to indicate
there are (virtually) no constraints on the second amino acid.
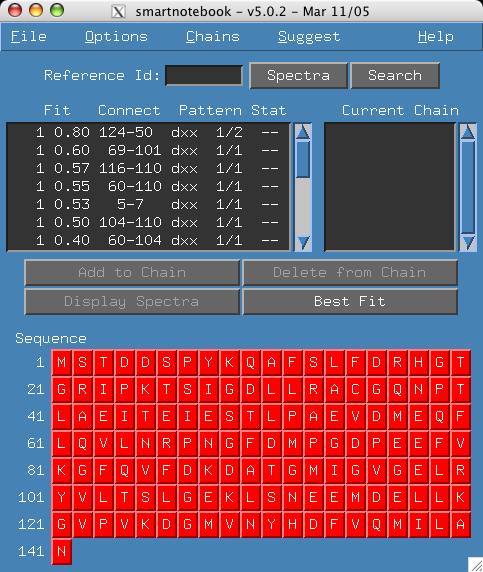
- Click Search in the search window. The software tells you there
are 17 hits and those
connectivities are displayed in the connections panel as shown below.
Example 2: I want to find connectivities that match a specific spot in
my sequence (eg, LYS 120 - GLY 121).
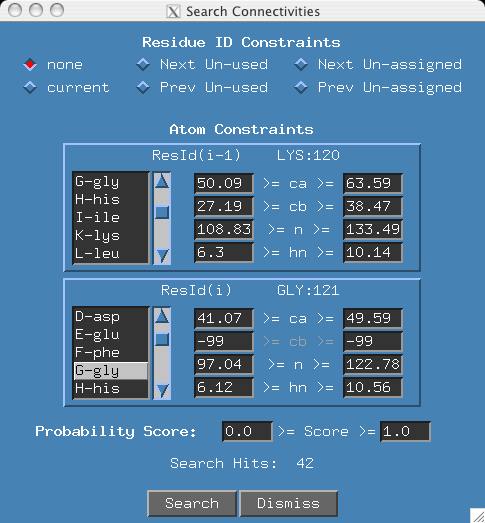
- In the ResId(i-1) window, click on the K-lys menu item until LYS:120
is displayed. In the ResId(i) window, click on G-gly menu item until GLY:121
is shown. Note that the 'cb' atom is not applicable and is darkened.
Click the "Search" Button and this time 42 connectivities are possible.
Note: Since most users will use default shift constraints files where
amino acids of a certain type have identical constraints, it is only
necessary to click on the desired amino acid once.
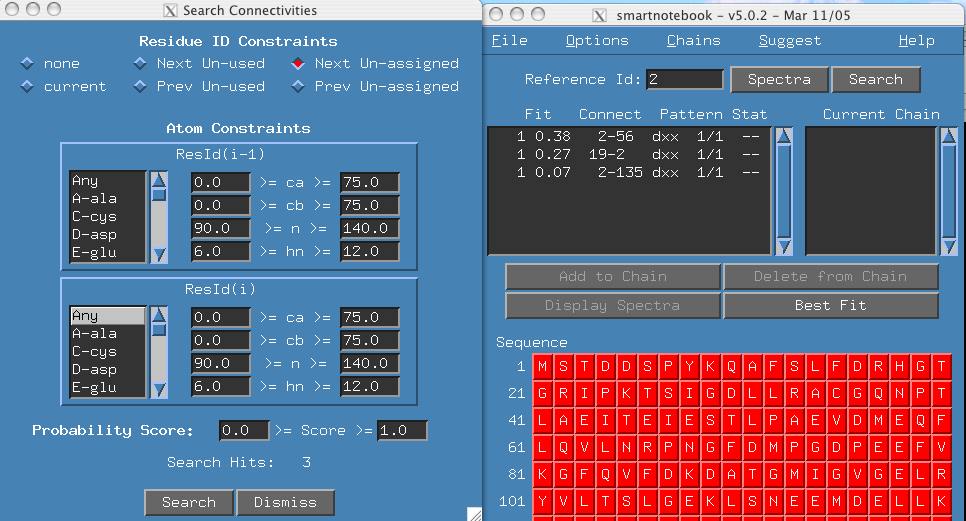
Example 3: How can I find out what connectivities have
not been assigned yet?
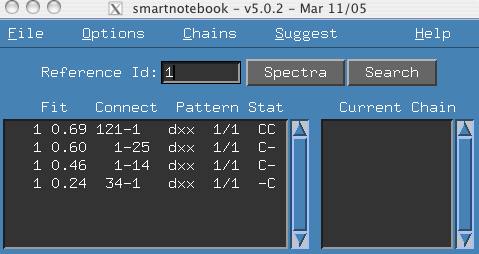
- (Optional) Set the "Reference Id" to 1. This means our current
set of connectivities involve reference ID 1.
- Click the Search button and select the menu item "Next Un-assigned".
Click the menu item "Any" for both Atom Constraints panels
and press "Search".
-
The output in the main snb panel indicates that Reference Id 2 is the
next unassigned reference peak and there are 3 possible connectivities.
As you continue to click on "Search" you quickly parse through your
connections looking for ones that look promising for chain building.
You can use a similar procedure to parse in the other direction
(eg, Prev Un-assigned ). Users may have a number of chains,
some of which are not yet assigned. So rather than looking for the
next un-assigned peak, it is more useful to see which reference peaks
are not found in any current chain (eg, Next Un-used button).
More Notes
- The lower and upper bounds of residue atom constraints are determined
by the value of the "stddev limit on assignment" which is shown whenever
you click the "best fit" button.
- The box 'Any' represents no shift constraints.
- Since all results from searches are sorted based on connectivity
score, it is not clear whether the Probability Score
constraint is necessary. It is possible that users may want to view
all connections that have some high minumum probability score and look at
these as possible starting points for building chains (eg, the
data is poor and you are only going to get limited assignment information).
- Users can manually set the constraint boxes. However you can define
your own
default atom constraints in the lib/experiments/*/*.search file.
The format is pretty straight forward.
It is probably best to edit a copy of the file in your current working
directory and then modify the snb.init file to point to the new
file location.
 Smartnotebook Home
Smartnotebook Home
This file last updated:
Questions to:
bionmrwebmaster@biochem.ualberta.ca