
|
PENCE / CIHR-Group
|

|
Funding for this software has been provided in part by the
Canadian Institutes of Health Research (CIHR Group)
and the
Protein Engineering Networks of
Centres of Excellence (PENCE).
|
|
PENCE / CIHR-Group
|
|
Funding for this software has been provided in part by the
Canadian Institutes of Health Research (CIHR Group)
and the
Protein Engineering Networks of
Centres of Excellence (PENCE).
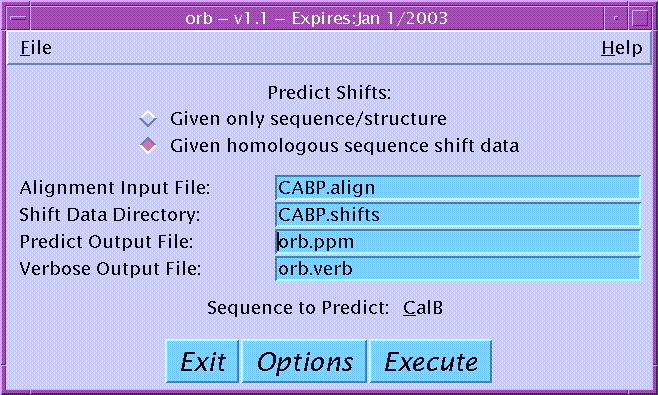
|
Latest Version: 1.2 - Dec 2002
Purpose: A program which predicts chemical shifts for a given sequence based on statistical analysis and/or previously assigned shifts of homologous sequences. |
The user puts all the homologus sequences into the xalign program to generate a sequence alignment file. The user then starts the orb program entering the sequence alignment file and the name of the directory containing the pertinent chemical shift files. The user selects the sequence to predict and selects from a group of options on the manner of the prediction. When the user hits the execute button, a prediction shift file (among others) is produced and user views the output by selecting the "Display Results" button.
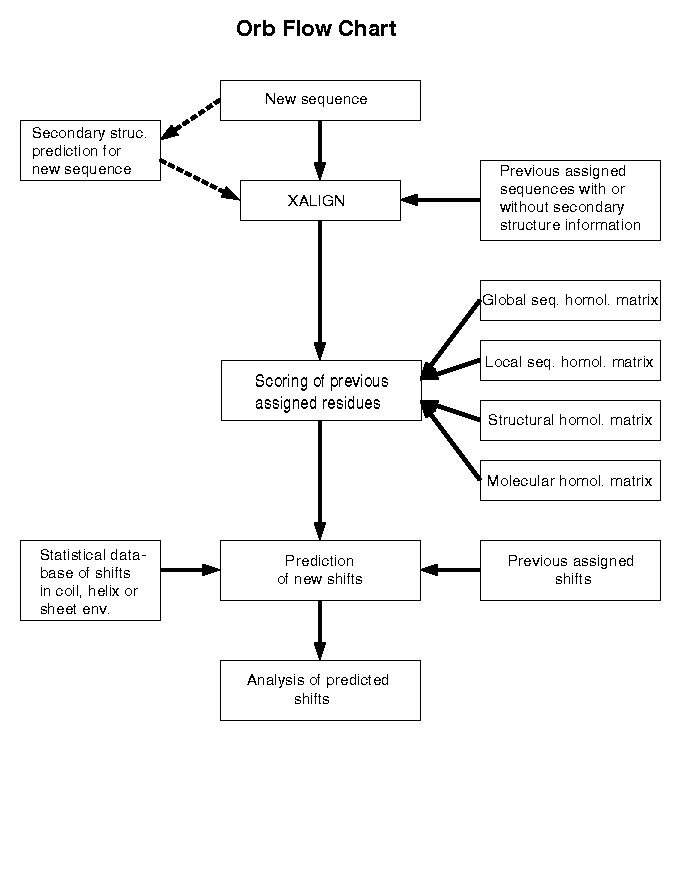
Click here to see a flowchart of the orb program.
Copyright (C) 1999 -
No portion of this program may be incorporated into other
programs or sold for profit without express written consent
of the authors.
PC(Linux): orb v1.2 (1.4 MB)
Once you have downloaded the software, you then proceed by
uncompressing and untarring the files. For example:
Do the following if you do NOT have any shift data from homologous sequences.
Create an input sequence file for your protein similar to the example below:
Orb will only use statistical database values for making predictions.
Do the following if you have shift data from one or more homologous sequences.
If you have installed orb, you can view an example homologous shift
directory in:
If you do not get a graphical window, check with your system administrator
to make sure the program has been installed and is accessible to you.
A common problem is that your PATH environment variable needs to
be changed to include the location of the installed orb program.
If you are logged in remotely, then enter the first command in the
console window and the second in your remote login window:
You can predict shifts given:
The program then displays the shifts directory field, the output
fields, and finally a menu which indicates the sequence to predict.
It is possible to get some pretty cryptic errors if you do not enter
a valid xalign output file at this point.
Hopefully you have remembered to put all your homologous shifts in
one directory. The naming convention for each shift file is "ID.PPM"
where ID is the sequence ID read in from the alignment file.
The program then displays an "Options" button.
You only need to do this if you do not like the default names
or you want to use a descriptive name which corresponds to the
sequence predicted.
See the "Orb output" section to learn about the output files
generated.
Note that orb selects the first sequence in the alignment file as
the default.
Sometimes you may want to experiment with different
combinations of shift files, or perhaps there are referencing
issues, or perhaps you have biases for one shift file over
another (which cannot be detected by using homology).
The program can take several seconds to run and produces several output
files in the current directory. The "Display Results" button appears
when the calculations are done.
If orb did not run cleanly, or the user aborts the gui prematurely,
all the temporary files (tmp.*) are kept around in the current
directory. These are likely not very useful to the average user and
should be removed.
General Algorithm
First it is necessary to obtain a multiple sequence alignment
of the new and homologous proteins. The alignment enables the
program to find all the homologous shift information which
pertains to any particular shift of the new protein.
Making a single shift prediction can simply be a matter of taking
a weighted average of the corresponding homologous shift data.
The weight for each homologous shift is currently determined by
the following factors:
Multiple Sequence Alignment
Because the multiple sequence alignment problem can be difficult
and subtle it was decided, for functionality sake, that
some other program should try to address this issue.
We chose the XALIGN program (Wishart et al., 1994) to accomplish
this task and designed orb to read the alignment output file.
A user can choose by some other method to create his/her own
alignment file provided it conforms to the XALIGN output format.
Weighting Homologous Shift Data
There are many factors we can consider in determining applicability
of homologous shift data used in predicting the new protein.
Currently orb uses a simplified set of criteria as explained
below where the specified variables are set in an
orb parameter file .
The program model assumes that proteins with higher homology
scores are considered more applicable for chemical shift prediction
than those with lower homology scores.
Global Sequence Homology
Each homologous protein is compared to the new protein and the
degree of primary sequence similarity determined.
Local Sequence Homology
Local sequence homology uses the same algorithm as global homology
except that the W is defined as W(i+n, i-n) where i is the residue
number of the current amino acid to predict and 2*n+1 defines the
window size.
Structural Homology
Structural homology considerations are limited to an assessment of
secondary but not tertiary structure homology. Secondary structure
is either known or can be calculated via structure prediction
programs (Chou & Fasman, 1974, 1978;...) or via the CSI index
(Wishart et al., 1992).
The calculation of the structural homology score is identical to the
local sequence homology score except a secondary structure similarity
weighting matrix is used instead of the amino acid silmilarity matrix.
Molecular Homology
Molecular homology describes the similarity between shifts arising from
residues that differ in type but have the same sequence position.
For example, one could use an assigned leucine ha to predict a
corresponding alanine ha in the new sequence via the following
formula:
Combining the Above Homology Factors
The next goal is to combine the above factors into a single
shift applicability score. The ORB programming model
uses the equation below:
Calculating the Predicted Shift
The following equation allows ORB to calculate a final predicted
shift s:
Non-stereo specific assignments
ORB can handle non-stereo specific assignments. First
we make predictions based on all applicable stereo specific data and
tables only, then
we modify our predictions based on the best way to fit the non-stereo
shifts to our predictions.
Orb is now smart enough to convert atom names of stereo specific
shifts to non-stereo specific as demonstrated in this example:
Non-homology Prediction Factors
So far orb can only make predictions based on homology. Sometimes a user
knows that a particular set of shifts may be more/less applicable
given the conditions of the experiment in which the shifts were
derived. By selecting the "Options" button you can increase/decrease the
shift bias multiplier for a given set of shifts. Then, once you
have hit "Execute", check the verbose output file to see how your bias
affects individual predictions. There is some amount of trial and error
here.
The shift name field has no blank characters and amino
acids are expected to have ResId's which are ordered
from lowest to highest.
A shift value field is specified as a either a real number or
with asterisks '*' to denote unknown values. The value "-999.99"
or "999.99" is also understood by several programs to mean an unknown value.
Here is an example of a typical PPM shift file:
This file last updated:
Questions to:
bionmrwebmaster@biochem.ualberta.ca
Copyright and Acknowledgements
Wolfram Gronwald ,
R. Boyko,
Frank Sonnichsen ,
David Wishart ,
and B.D. Sykes .
ORB, a homology-based program for the prediction of protein
NMR chemical shifts in J.Biomol.NMR 10, 165-181(1997)
Download
Select the version of orb corresponding to your operating system.
Solaris: orb v1.2 (1.4 MB)
SGI(Irix6.5): orb v1.2 (1.9 MB)
Installation
> uncompress orb-v1.2-sgi6.tar.Z
> tar xvf orb-v1.2-sgi6.tar
> cd orb-v1.2-sgi6
Look at the README file for details on installation.
> more README
It is pretty simple, all you have to do is know where you want to
put the executables and where to put the documentation, library and
example files. The installation script prompts you for
the names of these directories.
> ./Install
Finally you can test the program by going to the directory where the
program is installed and type the name. The README file also explains
how to set your path environment variable to include the location
of the executable.
Preparing Data Files
Although orb is a fairly easy program to use, there is a fair
amount of work in data preparation. Please carefully
follow the instructions below.
# This is an example sequence
>CaM Calmodulin - Drosophila melanogaster (1-148)
ADQLTEEQIAEFKEAFSLFDKDGDGTITTKELGTVMRSLGQNPTEAELQD
ccccchhhhhhhhhhhhhhccccccbbbhhhhhhhhhhcccccchhhhhh
MINEVDADGNGTIDFPEFLTMMARKMKDTDSEEEIREAFRVFDKDGNGFI
hhhhhccccccbbbhhhhhhhhhhhhhcccchhhhhhhhhhhhcccccbb
SAAELRHVMTNLGEKLTDEEVDEMIREANIDGDGQVNYEEFVTMMTSK
bhhhhhhhhhhcccccchhhhhhhhhhcccccccbbbhhhhhhhhhcc
Notes:
program on the input file above to generate an alignment
file. Make sure the multiple alignment results look reasonable.
>IL8.1a Interleukin 8 {1-72}
>IL8.H33A H33A Interleukin 8 Analog {6-72}
>TT Troponin-C III-III Homodimer {93-126,129-162}
Make sure each name only contains one set of braces and that your
amino acids in your corresponding shift files follow the numbering scheme
you have indicated.
$INSTALL/lib/orb/examples
Now you are set to run the program.
How to use Orb
xhost + remoteMachine
setenv DISPLAY hostMachine:0
This allows orb to run on the remote machine but the
display will go to the host computer.
Orb output
The output of orb consists of these files (assuming the default filenames):
How orb makes its predictions
This weighted average function becomes somewhat more complex
because we want to consider statistical shift database values
in the prediction when the homologous shift data is deemed
poor or is unavailable.
Finally, the program calculates a confidence interval for
each prediction based on the goodness of the homologous shift data.
gsh = x0 / xp * 100
A(ha) = L(ha) + (A(dbha) - L(dbha))
where A(dbha) and L(dbha) are average statistical values from the
Wishart database. The applicability of this converted shift information
is determined by the molecular homology table.
x(i) = sum all factors j (c(j) (y(j) - y0(j))
where
x(i) = applicability score for homologous shift i.
c(j) = coefficient for relative factor weighting. For example we
could choose to weight local homology more important than
global homology.
y(j) = score of a particular factor
y0(j) = minimum score for y(j). The advantage of including this
term enables x(i) to be < 0 identifying shifts which do
not meet a minimum criteria.
Any x(i) < 0 is set to 0 for convenience.
a0 * shift0 + sum all homologus shifts i (x(i) * shift(i))
s = -----------------------------------------------------------
a0 + sum all homologous shifts i (x(i))
where
a0 = weight assigned to database shift
shift0 = database shift value
x(i) = homologous shift applicability score
shift(i) = value of homologous shift i
Typically a0 is set to a small number in the ORB parameter file in
order to emphasize homologous shifts which exceed the minimum
applicability standards.
Essentially this is the ORB programming model. The researchers
have experimented with an exponential transformation on the x(i)s which
allows the best homologous shifts to get an even greater proportion
of weighting. An equation like a(i) = power(x(i), z) where z > 1 will
accomplish this.
1:ASP_32:HB1 3.10
1:ASP_32:HB2 3.10
is converted to
1:ASP_32:HB# 3.10
Other notes
$INSTALL/lib/orb/orb.parms
The orb.parms file is fairly well documented, read this file to get
an understanding for all the variables used in a prediction.
Appendix 1: PPM Formatted Shift Files
The following rules define a shift file in PPM format:
For example, 1:GLU_95:HB1 has molecular Number = 1,
Amino acid = GLU, Amino acid ID number = 95, and atom name = HB1
molNum:Residue_ResId:atom
where
molNum = Molecular Number (an integer)
Residue = Amino acid in 3 letter code (character string)
ResId = Amino acid ID number (an integer)
atom = Atom name (character string)
!
!Sequence: ADQ
!
1:ALA_1:N ***.**
1:ALA_1:C 174.00
1:ALA_1:CA 51.90
1:ALA_1:CB 18.80
1:ALA_1:HN ***.**
1:ALA_1:HA 4.15
1:ALA_1:HB# 1.57
1:ASP_2:N 120.50
1:ASP_2:C 175.80
1:ASP_2:CA 54.70
1:ASP_2:CB 41.20
1:ASP_2:HN ***.**
1:ASP_2:HA 4.67
1:ASP_2:HB1 2.72
1:ASP_2:HB2 2.60
1:ASP_2:CG **.**
1:ASP_2:HD2 *.**
1:GLN_3:N 119.60
1:GLN_3:C 175.80
1:GLN_3:CA 55.70
1:GLN_3:CB 30.20
1:GLN_3:HN 8.24
1:GLN_3:HA 4.42
1:GLN_3:HB1 2.12
1:GLN_3:HB2 2.00
1:GLN_3:CG 33.70
1:GLN_3:HG1 2.38
1:GLN_3:HG2 2.38
1:GLN_3:CD 180.00
1:GLN_3:NE2 ***.**
1:GLN_3:HE21 7.37
1:GLN_3:HE22 6.71
Appendix 2: Example orb.PPM output file
!
! Predicted shifts from orb
! Date: Fri Jul 4 14:16:46 1997
!
! Calcineurin B - human {1-170}
!
! Atom Predict Sdev RndCoil Sdev Confidence
!
1:MET_1:N 119.60 3.00 119.60 3.00 -
1:MET_1:HN 8.12 0.51 8.12 0.51 -
1:MET_1:CA 55.62 1.34 55.62 1.34 -
1:MET_1:HA 4.32 0.47 4.32 0.47 -
1:MET_1:CB 32.87 1.47 32.87 1.47 -
1:MET_1:HB1 1.84 0.84 1.84 0.84 -
1:MET_1:HB2 1.57 1.41 1.57 1.41 -
1:MET_1:HG1 2.20 1.23 2.20 1.23 -
1:MET_1:HG2 1.87 1.95 1.87 1.95 -
1:MET_1:HE# 1.47 1.73 1.47 1.73 -
1:MET_1:C 175.34 2.72 175.34 2.72 -
1:GLY_2:N 108.80 3.00 108.80 3.00 -
1:GLY_2:HN 8.36 0.71 8.36 0.71 -
1:GLY_2:CA 45.38 0.92 45.38 0.92 -
1:GLY_2:HA1 4.11 0.25 4.11 0.25 -
1:GLY_2:HA2 3.64 0.58 3.64 0.58 -
1:GLY_2:C 173.71 1.39 173.71 1.39 -
...
1:ALA_12:N 123.80 3.00 123.80 3.00 -
1:ALA_12:HN 8.11 0.71 8.11 0.71 -
1:ALA_12:CA 52.25 1.22 52.47 1.42 *
1:ALA_12:HA 4.17 0.29 4.19 0.34 *
1:ALA_12:CB 18.87 1.11 18.91 1.29 *
1:ALA_12:HB# 1.42 0.24 1.33 0.28 *
1:ALA_12:C 177.12 1.28 177.12 1.28 -
1:SER_13:N 115.30 2.71 115.70 3.00 *
1:SER_13:HN 8.20 0.59 8.30 0.64 *
1:SER_13:CA 58.03 1.35 58.10 1.48 *
1:SER_13:HA 4.42 0.23 4.38 0.28 *
1:SER_13:CB 64.11 1.02 63.86 1.19 *
1:SER_13:HB1 3.92 0.21 3.97 0.23 *
1:SER_13:HB2 3.82 0.30 3.83 0.33 *
1:SER_13:C 174.23 1.26 174.63 1.55 *
1:HIS_14:N 117.78 2.48 118.20 3.00 *
1:HIS_14:HN 8.30 0.53 8.36 0.63 *
1:HIS_14:CA 55.03 1.25 55.35 1.33 *
1:HIS_14:HA 4.70 0.31 4.59 0.38 *
1:HIS_14:CB 30.36 1.74 30.07 2.09 *
1:HIS_14:HB1 3.23 0.28 3.24 0.34 *
1:HIS_14:HB2 3.06 0.34 3.01 0.40 *
1:HIS_14:HD2 7.29 0.40 7.29 0.40 -
1:HIS_14:HE1 8.58 0.40 8.58 0.40 -
1:HIS_14:C 173.83 1.04 174.24 1.12 *
...
1:VAL_170:N 119.20 3.00 119.20 3.00 -
1:VAL_170:HN 8.12 0.68 8.12 0.68 -
1:VAL_170:CA 61.92 2.40 61.92 2.40 -
1:VAL_170:HA 4.12 0.44 4.12 0.44 -
1:VAL_170:CB 32.80 1.82 32.80 1.82 -
1:VAL_170:HB 2.06 0.23 2.06 0.23 -
1:VAL_170:HG1# 0.95 0.20 0.95 0.20 -
1:VAL_170:HG2# 0.81 0.23 0.81 0.23 -
1:VAL_170:C 176.04 1.54 176.04 1.54 -
Appendix 2: Example orb parameters file
**** Parameter file for orb ****
The following file contains all the parameters needed to run
orb. Orb is a program which tries to predict chemical
shifts for an unknown sequence given that the chemical shift
values for homologous sequences exist.
Parameter entries are preceded by 2 consecutive angle brackets.
You are expected to type in the appropriate parameter values
at this point (if you do not like the default values).
----------------------------------------------------------------
First create the amino acid database. To do this we need to
define the amino acids and the associated atom names.
Enter the file which specifies the amino acids and atom names.
>> $LIB/pep.def
----------------------------------------------------------------
Add the default chemical shift values for each amino acid atom
to the amino acid database. Orb uses the chemical shift
files compiled by David Wishart. The shifts are not all in
one file but are separated into carbon, proton and nitrogen
shifts.
How many files are there?
>> 3
Enter each file of chemical shifts.
>> $LIB/dsw.prot
>> $LIB/dsw.carb
>> $LIB/dsw.nitr
----------------------------------------------------------------
One of the factors in predicting chemical shifts is amino acid
homology. In general we can predict chemical shifts with
better accuracy in those regions of the alignment which are
more homologous.
Determining the degree of homology is done via a homology
scoring matrix.
Enter the location of the homology scoring matrix.
>> $LIB/wt.homology
----------------------------------------------------------------
Another factor in predicting chemical shifts is amino acid
structural homology. In general we can predict chemical shifts
with better accuracy in those regions of the alignment which are
more homologous in a secondary structure sense.
Determining the degree of homology is done via a homology
scoring matrix.
Enter the location of the structural homology scoring matrix.
>> $LIB/wt.structure
----------------------------------------------------------------
Another factor in predicting chemical shifts is determining
molecular similarity between amino acid shifts. This means
comparing shift values of atoms where amino acids are not
necessarily the same.
Enter the location of the molecular scoring file.
>> $LIB/mol.data
----------------------------------------------------------------
There are several criteria we must examine when predicting
chemical shifts (ie, statistical database values, sequence
homology, atom similarity between amino acids, etc). The
strength of these factors tells us how to weight the shift
information available in order to arrive at a prediction.
coef x(j) Window
>> 1.0 60 /* Molecular Homology */
>> 1.0 60 /* Global Amino Acid Homology */
>> 1.0 60 7 /* Local Amino Acid Homology */
>> 0.0 50 5 /* Local Structural Homology */
>> 5.0 /* Weighting for database score */
>> 1.0 /* Exponential term p1 */
**** End of Parameters list for orb ****
{kind=link}