| |
stc Brian Sykes Lab |
 |
| |
stc Brian Sykes Lab |
|
Version: 6.0.2 - Sep / 2011
Download and Installation
Purpose:
Stc - Structural Thermodynamics Calculations
A program which performs free
energy calculations from the structure
of different complexes.
STC performs free energy calculations from the structure of the different complexes in a graphical user interface. We have compiled executable versions for Mac and linux PCs.
In essence STC consists of two modules. The first module, calc_asa, calculates the change in accessible surface area (ASA) for the dissociation process from the coordinate files in the Brookhaven protein data bank (pdb) format using the algorithm ANAREA (Richmond, 1984) as implemented in the program VADAR (Wishart et al.,1994). The output files consists of the tabulated ASA of each atom of the complex, both the free forms of the enzyme and the ligand, as well the difference in ASA for each atom. The total changes in non-polar (all carbon atoms and sulfur atoms) and polar (all oxygen and nitrogen atoms) are summed up. In addition, the atomic change in ASA are regrouped per residues (and per side-chain) for calculating the change in S conf.
The module thermo calculates the energetics from the change in ASA. The total change in ASA is the sum of the ASAp and ASAnp which distinguishes between the contributions of polar and non-polar atoms. These are then used to calculate the change in Cp and Hod for a desired temperature T. From the change in ASA of the different side-chains involved in the dissociation, the conformational entropy gained for the ligand and the enzyme is calculated. Finally, the total entropy change is calculated as the sum of all the entropic contributions.
Acknowledgements: Thanks to Pascal Mercier who did an extensive test of stc 4.2 and provided many helpful suggestions. Thanks to Fred Richards and T.J.Richmond, whose accessible surface area fortran software is still used in stc. William Wilcox also did modifications on the addradii subroutine. The work of K.P. Murphy and E. Freire forms the basis for the thermodynamics programming section. Thermodynamics of structural stability and cooperative folding behavior in proteins. Adv Protein Chem. 1992;43:313-61.
Copyright (C) 1999 - No portion of this program may be incorporated into other programs or sold for profit without express written consent of the authors.
The recipient of the stc software† package agrees to the following:
†The term ``Software'' herein shall mean all computer programs, recompiled versions of these programs, derivative works, support material, documentation, manuals and databases pertaining to the stc package.
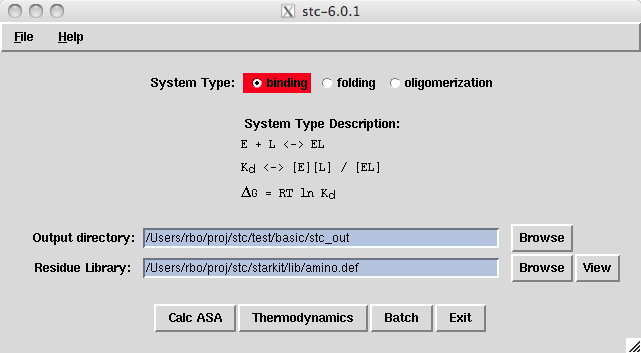
Note that the system type for our data is "binding". One could also choose "unfolding" or "oligomerization". See the examples.readme file for examples involving the other system types.
The output directory is where stc writes all the output files. If you start stc by clicking on the icon, the default output directory is $HOME/Desktop/stc_out. If you start stc via the command line, then the default output directory is ./stc_out. You can automatically set the output directory in the stc.defaults file or within the gui.
The residue library is where we define the amino acids, nucleotides, and parameters used in the calculations. Users with unique amino acids will need to create a customized residue library. You can automatically set the residue library in the stc.defaults file (see amino_def resource) or within the gui. See the xuw example we have provided.
The Calc ASA button allows one to calculate accessible surface area (ASA) output files. The Thermodynamics button lets the user make thermodynamic calculations based on the ASA files. Batch is a special case where the user wants ASA and thermodynamic calculations to be applied to several pdb data files.
Most users start by clicking on the Calc ASA to create the accessible surface area files. When that has been setup, press on the Thermodynamics button.
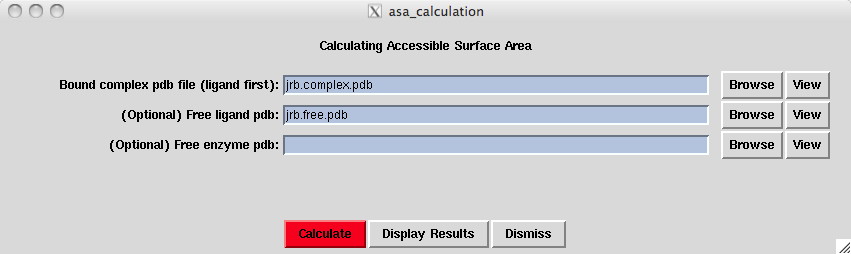
Because our system type is binding, we enter the bound complex pdb file and optionally enter the free ligand and free enzyme pdb files. Note: If the system type was "oligomerization", we enter the bound oligomer pdb and an optional free monomer pdb. For the "unfolding" system type, you enter both the folded and unfolded pdb files.
Click the Calculate button. The final message should say Done calculations, no errors. Otherwise click Display Results and check the stc.log file for clues on the errors.
Click the Display Results button.
A results window
When you are done, click on the
"Dismiss" buttons until you arrive back at the first stc window.
In this window we make thermodynamics calculations The Thermodynamics Window
based on accessible surface
area files. Note that stc fills in the "ASA file" fields
but the user can create or edit his/her own. The other fields are
set such that stc calculates sConf in a reasonable way.
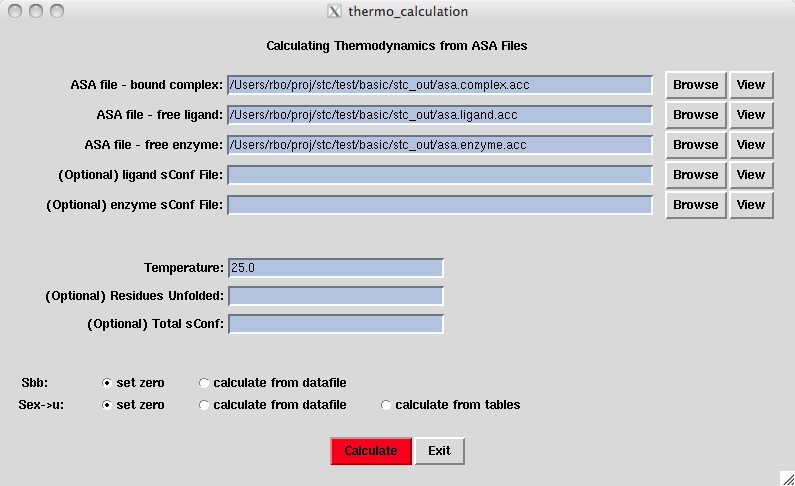
Although stc will calculate "sConf" automatically, the user has several options in determining how it is calculated. First note that calculating sConf for each residue is the summation of the individual entropy components "ΔSbu->ex", "ΔSex->u", and "ΔSbb". ΔSbu->ex is calculated from the amino acid table . ΔSex->u and ΔSbb are normally zero unless the user has specified residue ID numbers for residues that remain unfolded. In this case, these components are taken from the amino acid table.
Some users have experimental values for ΔSbb or ΔSex->u. In this case an optional sconf file allows the user to specify values of ΔSbb and ΔSex->u for each residue. You must click the "calculate from datafile" button in order to use these values or "estimate" which weights the ΔSex->u values according to total sidechain ASA of the molecule.
Click the Calculate button and see the next three sections for
a discussion of the results.
Section 1-ASA (Accessible Surface Area) values
This section defines the polar and nonpolar ASA values for
the free ligand, bound ligand, free enzyme, and bound enzyme.
These values are presented in a table 1. The
"free ligand ASA - bound ligand ASA" and
"free enzyme ASA - bound enzyme ASA" for both polar and nonpolar
quantities are what STC uses to calculate the thermodynamics of
binding. All changes in ASA are given in Section 2, Table 2.
ΔCpbind is the heat capacity of
binding and should be a negative value (positive if viewed as
dissociation). This value is calculated from the ΔASA nonpolar and
ΔASA polar values. The unit for heat capacity is kcal/mol/K.
ΔHbind is the enthalpy of binding and can be positive or
negative. If it is positive then the enthalpy of binding is
unfavorable, if negative then the enthalpy of binding is
favorable (the opposite is true for dissociation). The enthalpy of
binding is calculated from the heat capacity of binding plus
the binding enthalpy at a reference temperature of 60 degrees C.
The unit for enthalpy of binding is kcal/mol.
ΔSbind is the entropy of binding and can be positive or negative.
If this number is positive then the entropy of binding turns out to be
favorable, if negative then the entropy of binding turns out to be
unfavorable (the opposite is true for dissociation). The unit for
entropy of binding is kcal/mol/K. The entropy of binding is a sum of
3 entropies that are listed below. The first is the solvation entropy,
ΔSsol, which is directly calculated from the ΔASA nonpolar and
ΔASA polar values (unit is kcal/mol.K). The second is the overall
rotational/translational entropy, ΔSrt, which has a set value of -0.008
kcal/mol.K. The third is the conformational entropy, ΔSconf, which in
itself is a sum of 3 entropies. The first contribution to conformational
entropy is ΔSbu_ex, which is the change in conformational entropy of
the side chains due to tertiary or quaternary interactions during
binding. The second contribution to conformational entropy is ΔSex_u,
which is the change in conformational entropy of the side chains due to
secondary structure changes upon binding. The third contribution to
conformational entropy is ΔSbb, which is the change in conformational
entropy of the backbone upon binding. The 3 types of conformational
entropy are generally unfavorable with respect to binding resulting
in a negative value having the unit kcal/mol.K. The overall
conformational entropy of each residue is given in Table 4 in
the verbose output file. The individual
contributions (ΔSbu_ex , ΔSex_u, and ΔSbb) to conformational
entropy per residue is also given in Table 4 in the verbose output
file.
TΔSbind is the entropy of binding at the temperature
where the structure was determined. This value is useful in calculating
free energy.
ΔGbind is the free energy of binding and it is calculated from
ΔHbind - TΔSbind. A negative value for ΔGbind indicates binding is
favorable, a positive value for ΔGbind indicates binding in not
favorable. From ΔGbind one can quickly calculate a Ka and Kd. Many
of these thermodynamic values are given for each residue in Table
5 in the detailed output file.
Section 2: Individual Atom Differences in ASA
Section 3: Amino Acid Differences in ASA
Section 4: Calculating ΔSconf
Section 5-Thermodynamic values for each residue
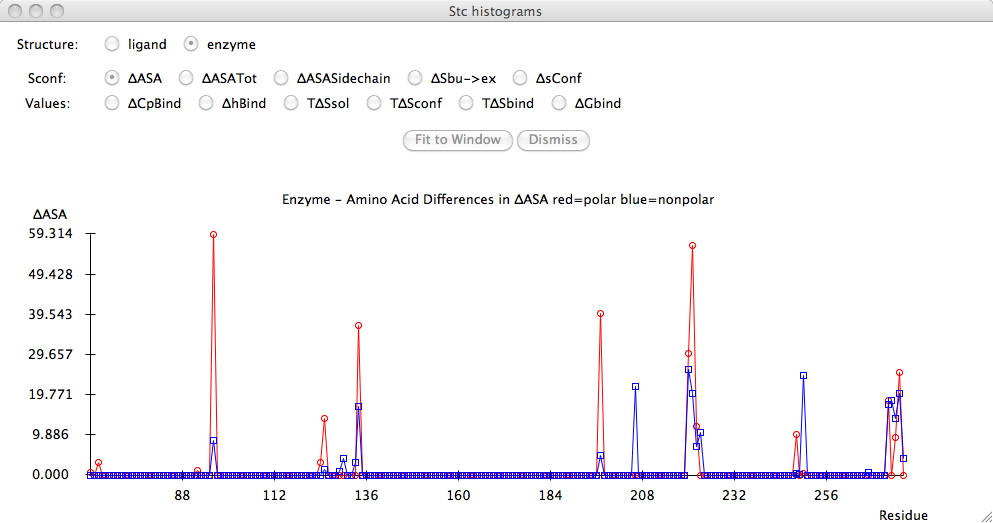
Notes: Unfortunately the histogram does not redraw automatically
when you resize the window, thus there is a "fit to window" button which
does it. Another note is that
the histogram does not know what to do when the residue Ids are
not unique. Probably a future version of this software will have
options to select the appropriate chain.
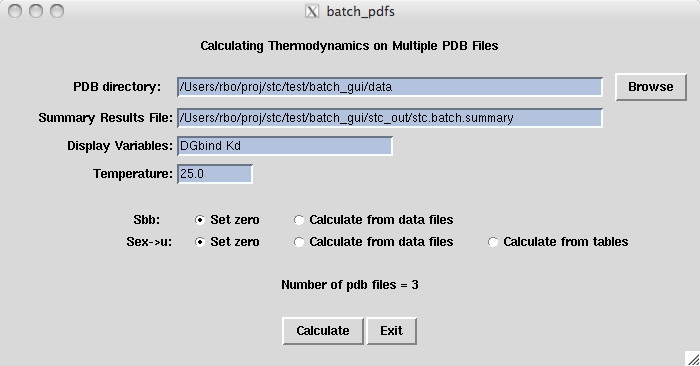
First you want to place all of your pdb files in one directory
as shown in our example data directory stc-examples/batch.gui.
In this example, the pdb files are made up of a ligand and an enzyme
separated by a TER statement. When you are ready, press the
Batch button on the main stc window.
Currently there are not alot of options, the most interesting being that
you can choose which variables you want to display as the results are
being calculated. Your choice of variables come from the typical
basic output file .
After pressing Calculate, each pdb file is processed in alphabetic order.
ASA output files are generated and thermodynamic calculations done with
default parameter settings.
A one-line summary of the thermodynamic results are shown on the screen and
sorted results are also available in the output file
stc.batch results . To see the thermodynamics
details for any particular pdf file, just click on the line containing the summary
results.
If you wish to bypass the gui altogether and are comfortable with writing scripts,
then take a look at the
batch_stc file in the examples directory.
Several users have expressed an interest in using routines within this package in
ways beyond the scope of this software.
Here are some notes for stc script writing.
Thermo is the independent
back-end of stc and a user could type thermo and be prompted for all
the input files required to perform the calculations. Thermo
produces two output files, a short version which stc displays
in a window and a verbose file.
Getting input files ready for thermo is a fairly complex
chore in itself. A shell script called calc_asa takes the
user's input pdb files and calculates the accessible surface area files
needed by thermo. The main program to do this is called access
which is a fortran program also used in
vadar.
Getting pdb files into acc format involves the following.
Here is a common problem in the stc.log file:
This file last updated:
Questions to:
bionmrwebmaster@biochem.ualberta.ca
Output - Basic results
The program displays a window which shows the
basic output file
of various binding constants and key variables.
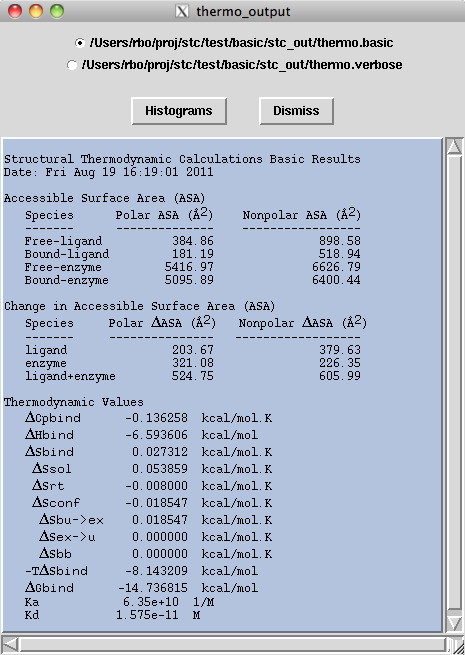
Table 1
Species Polar ASA (A2) Nonpolar ASA (A2)
Free Ligand # #
Bound Ligand # #
Free Enzyme # #
Bound Ligand # #
Section 2-Change in ASA values
This section defines the change in ASA, nonpolar and polar, when
comparing the free verses bound ligand and free verses bound enzyme.
The difference in polar area upon binding for the ligand is then
added to the respective polar value for the enzyme. This total
change in ASA polar is what STC directly uses for thermodynamic
calculations. The difference in nonpolar area upon binding for the
ligand is then added to the respective nonpolar value for the enzyme.
This total change is ASA nonpolar is what STC directly uses for
thermodynamic calculations. These are global values, the ΔASA for
individual atoms and residues are listed in the detailed output
file Tables 1-4.
Table 2
Species Polar ΔASA (A2) Nonpolar ΔASA (A2)
ligand 222.30 740.50
enzyme 513.14 770.96
ligand+enzyme 735.44 1511.46
Section 3-Thermodynamic values
This section defines the thermodynamic values calculated from the ΔASA
nonpolar and ΔASA polar contributions from the ligand + enzyme. The
thermodynamic calculations listed are the global values, the individual
atom or residue values are listed in the detailed output file. For more
details please look up our references and acknowledgements.
Output - Verbose results
If you click on the
verbose output file
you can see thermodynamic output on specific atoms or residues.
Here is where you can study the individual atom differences,
the classification of polar/nonpolar atoms, a residue by
residue summary in the calculation of sconf (including the
change in ASA for total polar and non-polar atoms) and
the calculation of themodynamic paramaters per residue.
This section displays the results for atoms having a change in their
ASA when comparing the free ligand with the bound ligand and free
enzyme with the bound enzyme. The first table shows the results for the
ligand, the second table is for the enzyme. Only atoms with a
change in ASA are shown.
ResId Name Atom ΔASA Definition
A2
1 ALA N 0.2363 polar
1 ALA CA 11.3143 nonpolar
1 ALA C 0.1231 nonpolar
1 ALA O 0.1229 polar
1 ALA CB 12.8967 nonpolar
2 LEU CA -0.3348 nonpolar
2 LEU C 0.5131 nonpolar
...
This section displays the results for residues having a change in their ASA
when comparing the free ligand with the bound ligand and free enzyme with
the bound enzyme. The first table shows the results for the ligand, the second
is for the enzyme. Only residues with a change in ASA are shown.
ResId Name ΔASA Polar ΔASA Nonpolar
A2 A2
64 ASP 0.8498 0.0000
66 HIS 3.2483 0.0000
92 ASP 1.3114 0.0000
96 ARG 59.3139 8.5508
124 ASN 3.2231 0.0000
...
This section displays the calculation for the conformation entropy
for each residue. This is done separately for the ligand and the
enzyme. It also includes the ΔSbu->ex, ΔSex->u, and ΔSbb values used
to compute ΔSconf per residue. ΔASATot is the sum of nonpolar ΔASA
and polar ΔASA for each residue. ΔASAsc is the change in ASA for the
side chain of each residue. The
next 3 entropy values deal with
structural changes in side chain or backbone conformations upon binding.
When added together they total ΔSconf.
ResId Name ΔASATot ΔASAsc ΔSbu->ex ΔSex->u ΔSbb ΔSconf
A2 A2 cal/mol.K cal/mol.K cal/mol.K cal/mol.K
1 ALA 24.6933 12.8967 0.0000 0.0000 0.0000 0.0000
2 LEU 68.4463 68.0124 0.8092 0.0000 0.0000 -0.8092
3 MAS 26.6067 26.7675 0.0000 0.0000 0.0000 0.0000
...
This section gives the heat capacity of binding, the enthalpy of binding,
the different entropies of binding, and the free energy of binding for each
residue. The entropy values are multiplied by the temperature the
structure was calculated at (oK).
ResId Name ΔCpbind ΔHbind TΔSsol TΔSconf TΔSbind ΔGbind
kcal/mol.K kcal/mol kcal/mol kcal/mol kcal/mol kcal/mol
1 ALA -0.0109 0.5741 0.8327 0.0000 0.7581 -0.1840
2 LEU -0.0306 1.6392 2.3402 -0.2413 2.0244 -0.3852
3 MAS 0.0041 -0.8211 -0.0691 0.0000 -0.1437 -0.6774
4 ARG 0.0046 -1.8806 0.3116 -0.6848 -0.4477 -1.4328
...
Output - histograms
Finally, in order to analyze this output more easily, you can
click on the "Histograms" button where you can receive graphical
displays of the
key thermodynamic indicators
residue by resiude. Clicking on the radio button automatically
draws the requested histogram.
Multiple Structures
You have several structure files and you want to see which one
has the most favorable thermodynamics. This window allows the user
to do this quickly.
Multiple Structure Scripts
Troubleshooting
*** Error, Unrecognized residue name: 577 S1 EMD 1 61.88000
1.41900 0.21200 1.8000
Define this residue in the amino.def file if vanderwaal assumptions are wrong
One or more user pdb files contains an amino acid that STC does not
recognize. See the stc-examples/xuw directory where there is an
example
of how to modify the amino.def file.