> cd tutorial > ls > stc

Note that the system type for our data is "binding". One could also choose "unfolding" or "oligomerization". Our examples library contains sample runs of these system types.
The output directory is where stc writes all the output files. The residue library is where we define the amino acids, nucleotides, and parameters used in the calculations. Users with unique amino acids will need to create a customized residue library. The directory lib/examples/xuw is an example of this type of problem.
The Calc ASA button allows one to calculate accessible surface area (ASA) output files. The Thermodynamics button lets the user make thermodynamic calculations based on the ASA files. Batch is a special case where the user wants ASA and thermodynamic calculations to be applied to several pdb data files.
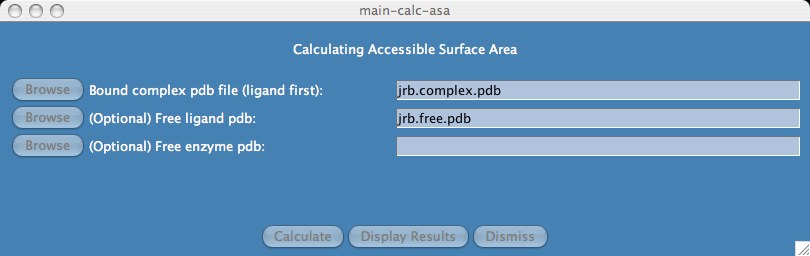
Because our system type is binding, we enter the bound complex pdb file and optionally enter the free ligand and free enzyme pdb files.
Note: If the system type was "oligomerization", we enter the bound oligomer pdb and an optional free monomer pdb. For the "unfolding" system type, you enter both the folded and unfolded pdb files.
- Click the "Calculate" button.
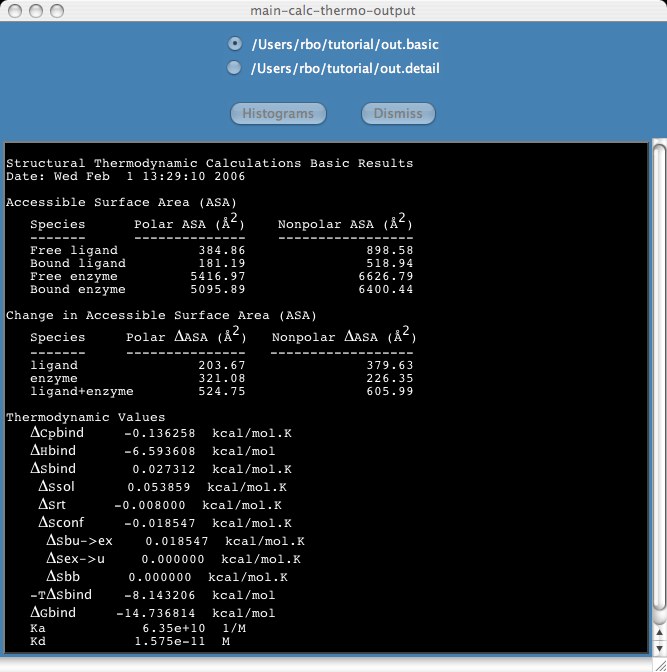
The program prints a number of messages to the screen telling you what it is doing. The final message should say "Done calculations, no errors".
- Click the "Display Results" button.
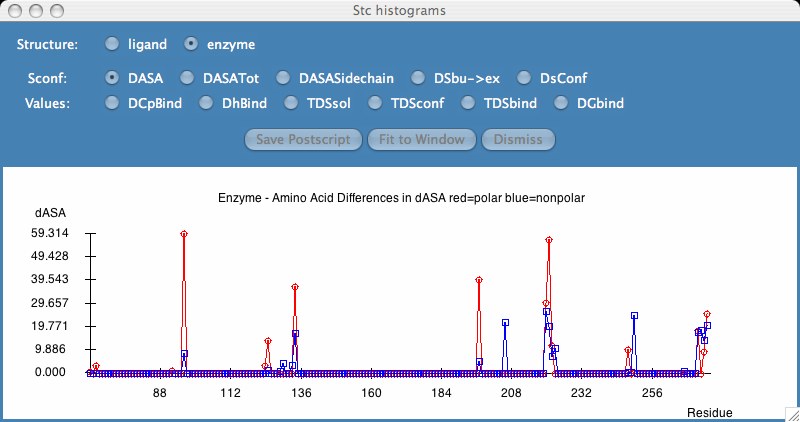
A results window