 |
xalign Brian Sykes Lab |
 |
| |
xalign Brian Sykes Lab |
|
|
Version: 6.1 - Apr 2009
Purpose: a graphical X-windows program for multiple sequence alignment based on sequence homology and secondary structure. |
xalign has all of the following attributes which make it a very powerful yet relatively easy to use program:
Copyright (C) 1994 - No portion of this program may be incorporated into other programs or sold for profit without express written consent of the authors.
Linux:
Once you have downloaded the software, you then proceed by
uncompressing and untarring the files. For example:
If you do not get a graphical window, check with your system administrator to
make sure the program has been installed and is accessible to you. A
common problem is that your PATH environmental variable needs to be
changed to include the location of the installed xalign program.
If you are logged in remotely, then enter the first command in the
console window and the second in your remote login window:
See the Input Files section for more
information.
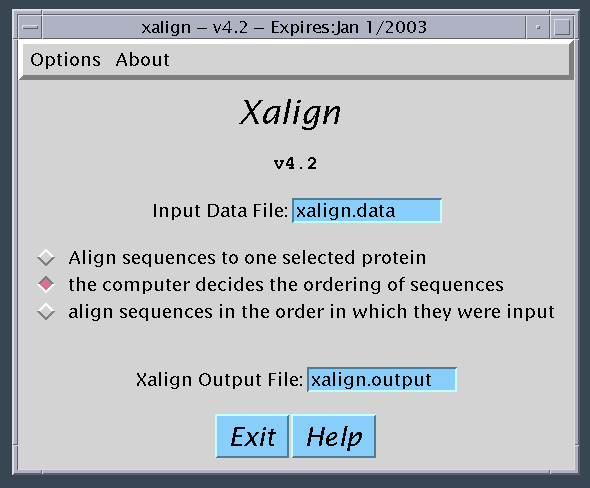
If you click the first button, the sequences are displayed and the user
clicks the sequence to align to.
Since the multiple sequence alignment algorithm is heuristic, xalign
can generate different alignments depending on the order in which
the sequences are processed.
The default computer algorithm is to align
sequences from most to least homologous,
starting first with those sequences that have structure determined.
You as the user have the choice of selecting the initial sequence to
align to or even deciding the complete order for processing sequences.
This freedom is basically allowed for experimental purposes. Most of
the time your best alignment should occur when you select the option
that allows the computer to decide the alignment order.
Each input sequence must contain these minimum attributes:
The number of amino acid codes per line does not matter,
however, it is easier to check your input for correctness if
you decide on a constant number such as 50. Also, blanks are
ignored if found in the amino acid sequence. Alternative
amino acid code meanings such as 'B', 'X', and 'Z' are
acceptable input but they will have no scoring value during
the alignment process (unless the amino acid scoring matrix
is changed).
Here is an example input file:
Here is an example input file with secondary structure
information included:
The following section explains how the user can enter
specific knowledge into the alignment process.
Sometimes xalign will insert gaps into an alignment where
you think are not correct. You could change any of the various
gapping penalties in the parameter file but this will
likely change your entire alignment (which you may already
be happy with). To prevent the program from breaking up a
certain section of amino acids, just type asterisks above
those amino acids. Because the program ignores blanks in
the input sequence, the other amino acids without asterisks
must get some kind of default character. In this case, use a
"-" or dash character.
Here is an example of a sequence input file which has this amino
acid clustering:
Another potentially useful tool is to be able to anchor a
certain amino acid in one sequence to a certain amino acid
in another sequence. One can imagine a scenario where a user
knows that two amino acids line up but because of remote
homology, xalign can't understand the significance of that
particular match.
To implement this anchoring procedure, the user specifies a
number between 1-5 above the first amino acid to anchor in
the first sequence. The user then specifies that same
number above the second amino acid in the second sequence.
Here is an example of anchoring one amino acid in one
sequence to another amino acid in another sequence:
The printing of pairwise alignments is an option for the
user in the
xalign.parms
file. The bars "|" in the pairwise
alignment indicate amino acids which are identical, the
asterisks "*" denote amino acids which are similar. The
currect amino acid number is printed at the end of each
sequence line.
The "percent sequence homology" is calculated as the score of the
current alignment divided by the score of the perfect alignment.
It is not the number of amino acids which match over the length
of the alignment.
The ranking of pairwise sequences is determined by the
"alignment score". The alignment score
is the percent sequence homology score plus a constant if the sequence
has secondary structure determined.
The order of sequences in the multiple
alignment is based on the sequences which occur first within
the ranked pairwise alignments. Usually it is better to
align sequences from most to least homologous starting first
with those that have structure determined.
Note that the consensus sequence is based on a threshold
percent identity which is set in the parameter file. If the
threshold is reached, then that amino acid is printed otherwise
a dash "-" is printed.
The xalign program was developed to handle the majority of alignment
requests in a reasonable manner. Compromising the relatively straight
forward algorithm for special classes of alignments of probable nature
seemed beyond the intent of the program. Since the tools are available for
the user to correct the errors, let he/she use them.
The following suggestions can help you use the xalign
program to arrive at the best alignment possible. Some
of these suggestions involve modifying variables (denoted as XALIGN:)
in the
xalign.parms
file. There are also interesting paramters in
wtm.rbo .
You should copy these two files from the xalign lib directory to your
current directory and make appropriate changes.
The "xalign.parms" parameter file contains default settings for
gap penalties, amino acid similarity and also some useful output options.
The program looks for an "xalign.parms" file in the current directory, if
one does not exist it uses the xalign.parms file found in the
directory where you installed the program.
Users who are interested in changing some of default
settings in order to get better alignments may want to copy
the above file to their current directory and try various
changes.
An example parameter file is shown below.
This file last updated:
Download
Select the version of xalign corresponding to your operating system.
Solaris:
Intel Mac (aqua)
Intel Mac (x11)
Powerpc Mac (aqua)
Powerpc Mac (x11)
Installation
> uncompress xalign-v6.1-linux.tar
> tar xvf xalign-v6.1-linux.tar
> cd xalign-v6.1-linux
> ./Install
For Mac aqua versions, after untarring (or double clicking the download) drag the
xcrvfit icon to /Applications.
Running xalign
xhost + remoteMachine
setenv DISPLAY hostMachine:0
This allows xalign to run on the remote machine but the
display will go to the host computer.
Basic Sequence Input File for xalign
An input file contains two or more sequences to align.
Although there is no maximum number of sequences you can align,
you are limited by the amount of memory on the machine you
are running on.
>CaM Calmodulin - Drosophila melanogaster (1-148)
ADQLTEEQIA EFKEAFSLFD KDGDGTITTK ELGTVMRSLG QNPTEAELQD
MINEVDADGN GTIDFPEFLT MMARKMKDTD SEEEIREAFR VFDKDGNGFI
SAAELRHVMT NLGEKLTDEE VDEMIREANI DGDGQVNYEE FVTMMTSK
>TnC Troponin C, cloned chicken skeletal muscle (1-162)
ASMTDQQAEA RAFLSEEMIA EFKAAFDMFD ADGGGDISTK ELGTVMRMLG
QNPTKEELDA IIEEVDEDGS GTIDFEEFLV MMVRQMKEDA KGKSEEELAN
CFRIFDKNAD GFIDIEELGE ILRATGEHVI EEDIEDLMKD SDKNNDGRID
FDEFLKMMEG VQ
To include secondary structure in a sequence, this information
is placed on the line directly below the primary
sequence (upper or lower case letters acceptable). Use "h"
for helical regions, "b" for beta strand, "c" for random
coil, "t" for beta turn and "x" for regions you don't know
or care about.
>CaM Calmodulin - Drosophila melanogaster (1-148)
ADQLTEEQIAEFKEAFSLFDKDGDGTITTKELGTVMRSLGQNPTEAELQD
ccccchhhhhhhhhhhhhhccccccbbbhhhhhhhhhhcccccchhhhhh
MINEVDADGNGTIDFPEFLTMMARKMKDTDSEEEIREAFRVFDKDGNGFI
hhhhhccccccbbbhhhhhhhhhhhhhcccchhhhhhhhhhhhcccccbb
SAAELRHVMTNLGEKLTDEEVDEMIREANIDGDGQVNYEEFVTMMTSK
bhhhhhhhhhhcccccchhhhhhhhhhcccccccbbbhhhhhhhhhcc
>TnC Troponin C, cloned chicken skeletal muscle (1-162)
ASMTDQQAEARAFLSEEMIAEFKAAFDMFDADGGGDISTKELGTVMRMLG
cccchhhhhhhhhcchhhhhhhhhhhhhhccccccbbbhhhhhhhhhhcc
QNPTKEELDAIIEEVDEDGSGTIDFEEFLVMMVRQMKEDAKGKSEEELAN
cccchhhhhhhhhhhccccccbbbhhhhhhhhhhhhhcccccccchhhhh
CFRIFDKNADGFIDIEELGEILRATGEHVIEEDIEDLMKDSDKNNDGRID
hhhhhccccccbbbhhhhhhhhhhhccccchhhhhhhhhhhccccccbbb
FDEFLKMMEGVQ
hhhhhhhhhhcc
Note: If you choose to enter secondary structure information,
then you must enter it for all amino acids.
Advanced Sequence Input File for xalign
>unkn1 unknown protein mouse
-------------******-------------------------------
SRTEYDPLKFWPITHYCPHSARKDTYPERFYANMPKLDNQGPLSTYPLST
cchhhhhhhhhchhhhhhhccccccccccbbbbccccccccccccchhhh
---------------
QWPIIVDTASATLMS
hhcbbbbbbcccccc
In the example above, the asterisks over "THYCPH" will
ensure that the program will not break up these amino acids
in the alignment.
>unkn1 this protein unknown for mouse
---------1----------------------------------------
SRTEYDPLKFWPITHYCPHSARKDTYPERFYANMPKLDNQGPLSTYPLST
cchhhhhhhhhchhhhhhhccccccccccbbbbccccccccccccchhhh
---------------
QWPIIVDTASATLMS
hhcbbbbbbcccccc
>unkn2 some other protein
---------1----------------------------------------
SRSELDPLKFMPLPITYCGHSAREATYPERDDANMPKLENSTGPLQTYPL
------------------
LSYQCPIIVDTAKHLLNS
The anchoring procedure can be applied to any number of
sequences. You can also have the same anchoring number
appear more than once in a sequence, the program ends up
choosing the anchor which maximizes the total alignment score.
Output Files
The output of xalign consists of the following:
Analyzing your Alignment
First it is important for the user to realize that the programming
model makes a number of assumptions and simplifications in order
to turn multiple sequence alignment into a mathematical problem.
Secondly, the user should realize that solving this particular
mathematical problem "perfectly" is impractical for
3 or more sequences.
> cp /usr/local/lib/xalign/xalign.parms .
> cp /usr/local/lib/xalign/wtm.rbo .
> vi wtm.rbo
XALIGN:SCOR_MATRIX wtm.rbo
Alignment Parameters
****************************************************************
XALIGN Parameters
****************************************************************
Enter the file which contains the amino acid similarity
scoring matrix and the various gap penalties.
This file contains a lower triangular matrix which indicates
the similarity score between two amino acids. The matrix is
of a general nature and may not reflect the specific needs
of the user (for example, a user who finds that cystines are
not lining up with cystines may want to increase the score
for such a match).
XALIGN:SCOR_MATRIX $INSTALL/lib/xalign/wtm.rbo
----------------------------------------------------------------
Indicate if pairwise alignments should be printed. This can
greatly increase the output however it often contains useful
information. Enter "1" for printing and "0" for not printing.
XALIGN:PRINT_PAIRWISE 0
----------------------------------------------------------------
The following is an offset added to the percent homology score
of a pairwise alignment if secondary structure is known for a
given sequence. In general, any sequence which has secondary
structure known should be given precedence over those that do not
when determining the order of sequences in the multiple alignment.
An offset of "0" means the order of the sequences in the multiple
alignment algorithm is based only on primary sequence homology.
Range: MIN_STRUCT_BIAS <= x <= MAX_STRUCT_BIAS
XALIGN:STRUCT_BIAS 100
----------------------------------------------------------------
As more and more sequences are added to the multiple alignment,
the cumulative effect of all the amino acid scoring starts to
dwarf the gapping penalties. Therefore we probably want to
increase the gap and gap size penalties by a given percent for
each sequence added.
For example, this number should be adjusted up if too many gaps
are found in the final sequences of the multiple alignment.
Range: MIN_INC_GAP_PEN <= x <= MAX_INC_GAP_PEN
XALIGN:INC_GAP_PEN 30
----------------------------------------------------------------
Indicate if the pairwise sequence percent homology table should
be printed.
This table allows the user to quickly compare pairwise sequence
alignments in terms of percent homology.
Enter "0" for not printing and "1" for printing.
Range: 0 <= x <= 1
XALIGN:PRINT_PCT_HOM 1
----------------------------------------------------------------
Indicate the order of the sequences in your multiple alignment
output.
To have sequences printed in the order that the program ranks
them (for adding them to the multiple alignment), enter "0".
If you prefer to have the sequences printed in the order that
they were entered into the program, enter "1".
Range: 0 <= x <= 1
XALIGN:PRINT_ORDER 0
----------------------------------------------------------------
Following the multiple alignment, a consensus sequence is
printed. At what percent identity is a consensus reached for
a given amino acid at a given position?
Enter an integer from 0 - 100 to represent this threshold.
Range: 0 <= x <= 100
XALIGN:CONSENSUS_PCT 70
----------------------------------------------------------------
Do not change these parameters, they were added for the sake of
compatibility with other packages that use the low level
xalign engine.
XALIGN:DATA_FORMAT 1
XALIGN:DATA_FILE x
XALIGN:SEQ_SELECT 1
Default Weighting Matrix
#
# rbo amino acid similarity matrix
#
# This matrix indicates how important it is to line up any given
# pair of amino acids (eg, aligning a cysteine with another
# cysteine is worth "n" points). 20 refers to number of amino
# acid character codes.
KEY:WT_PMAT 20
A C D E F G H I K L M N P Q R S T V W Y
A 10
C 2 17
D 2 2 10
E 2 2 6 10
F 1 0 0 0 12
G 2 2 2 2 0 10
H 3 1 2 2 3 2 10
I 2 2 0 0 6 0 1 10
K 3 2 3 3 0 2 3 0 10
L 2 2 0 0 6 0 0 4 0 10
M 2 2 0 0 4 0 2 4 1 8 10
N 2 0 6 4 0 3 3 0 7 0 1 10
P 2 0 3 0 0 2 3 0 2 0 0 2 10
Q 2 0 4 6 0 1 4 1 4 0 1 6 2 10
R 2 2 2 2 0 2 4 0 7 0 1 2 2 4 10
S 5 2 2 2 0 2 1 0 2 0 0 4 3 2 3 10
T 3 2 2 2 0 2 2 2 2 2 2 2 2 2 2 4 10
V 2 2 0 0 3 1 1 8 0 6 4 0 0 0 0 0 2 10
W 0 0 0 0 5 0 1 0 0 1 1 0 0 1 3 1 0 0 16
Y 0 1 0 1 9 0 2 3 0 2 3 0 1 1 0 1 0 3 5 12
#
# This matrix indicates how important it is to line up any given
# pair of amino acid structure types.
#
# B = betaStrand
# C = random coil
# H = helical
# T = beta turn
# X = don't know or don't care
#
KEY:WT_SMAT 5
B C H T X
B 3
C -1 2
H -3 -1 3
T 0 -1 -1 2
X 0 0 0 0 0
#
# If this matrix is used in sequence alignments, what would be
# the penalty for introducing a gap in the alignment. Increasing
# this number reduces the overall number of gaps in your alignment.
KEY:GAP_PEN 10
#
# Enter the penalty for each blank in the gap (ie, gap size).
# Should a big gap be penalized more than a small gap? In most
# cases, yes. However this penalty can be zero or some small
# number if the sequences are roughly the same length. Increase
# this penalty if your alignment contains large unrealistic gaps.
KEY:GAP_PEN_SIZE 2
#
# In the above smiliarity matrix specify the score that would constitute
# a near match between amino acids. This is used for printout purposes.
KEY:NEAR_MATCH 5
#
# Indicate the penalty for inserting a gap in the middle of a
# helix or beta strand structure (if known).
#
# Note that this is a penalty in addition to the regular gap
# penalty as described above.
KEY:MID_GAP_PEN 30
#
# Indicate the penalty for inserting a gap near the end of a
# helix or beta strand structure.
#
# Typically this value is 3 or 4 times less than the middle of
# structure penalty directly above.
KEY:END_GAP_PEN 10
#
# The above similarity matrix does not indicate the score of matching
# an amino acid with a blank (eg, a gap in an alignment). Matching a
# blank with an amino acid in a multiple alignment is not necessarily
# a bad thing (particularly gaps before or after a sequence).
KEY:BLANK_MATCH 0